Inhibitor for Human Galectin 3 (PDB ID 3ZSJ)
Galectins are carbohydrate-binding proteins serving as regulators in many essential cell processes: polarity, adhesion and motility, metastasis, apoptosis, nuclear and immune functions. Due to such a rich role in the animals, galectins are important drug targets. Most of the galectin inhibitors are based on carbohydrates, their natural ligands. Therefore, there is a significant interest amongst pharmaceutical community to develop a non-sugar based galectin inhibitor. In this section we have put an example of how this could be achieved using our algorithm.
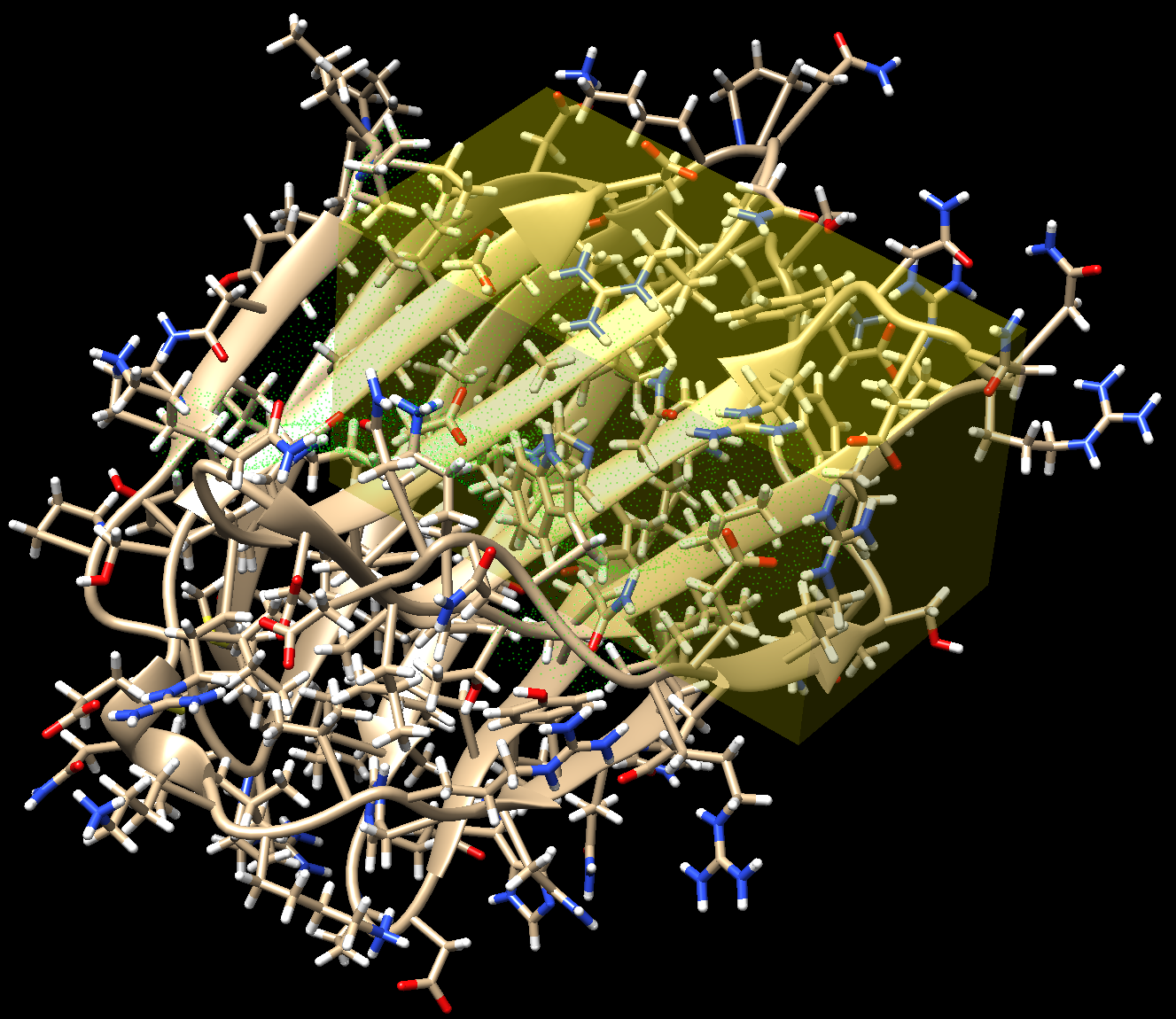
First, the target macromolecule and the binding site are identified. The binding site is defined as a parallelepiped zone where the search for the ligand structure is to be conducted (Fig. 1).
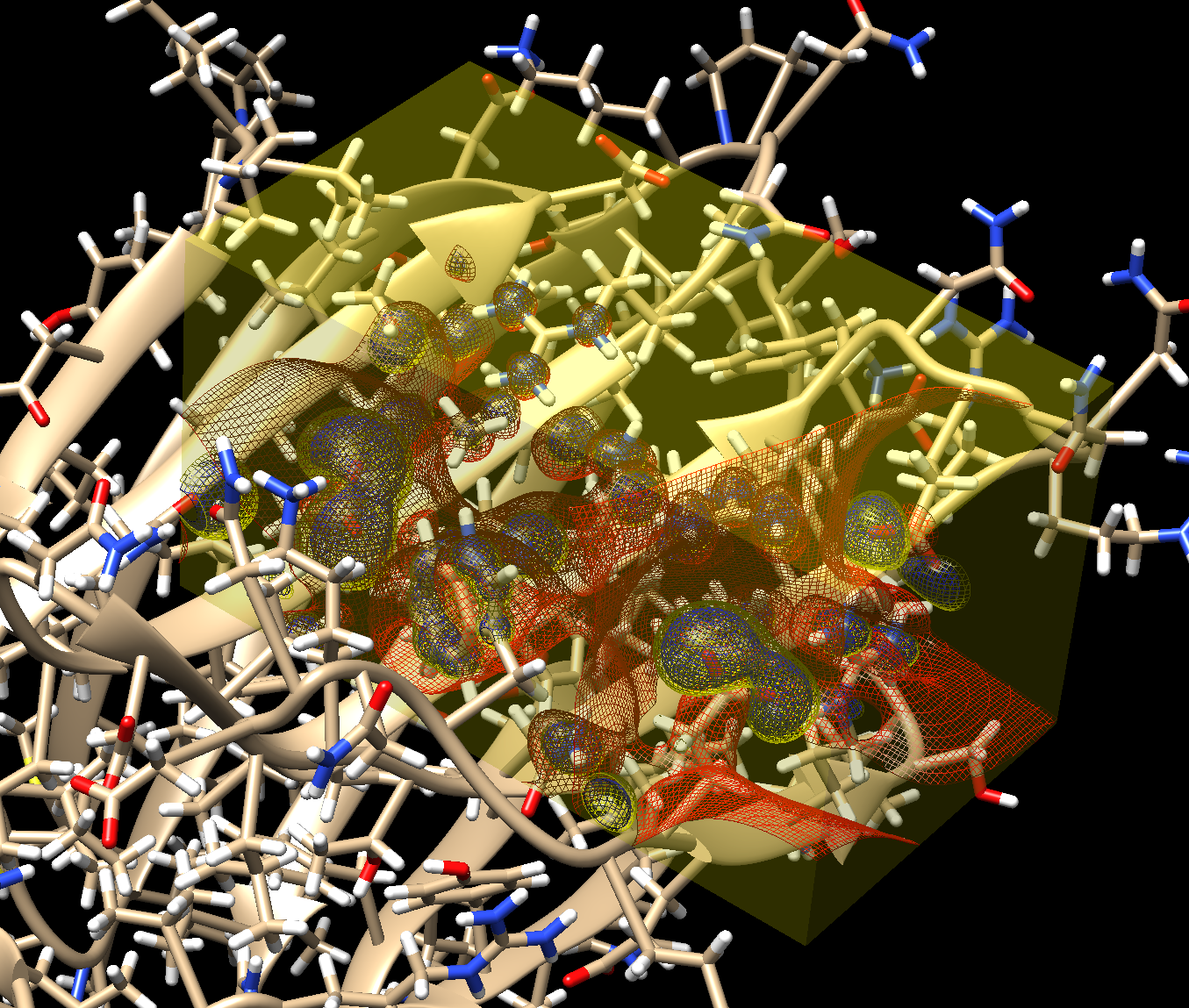
Then the electrostatic potential of the target molecule is calculated solving the Poisson-Boltzmann equation (Fig. 2).
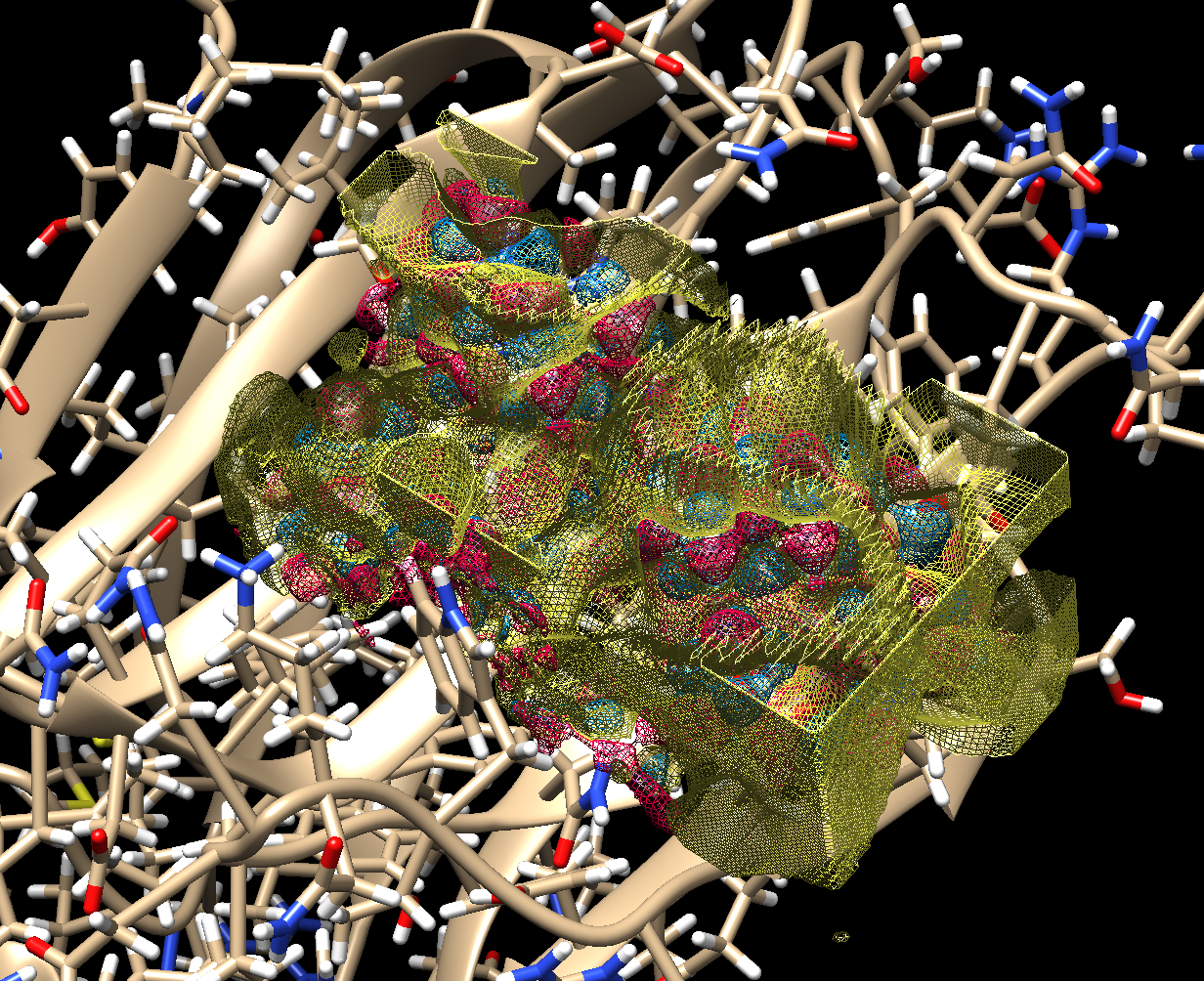
This is all what is required for the algorithm to proceed to calculate the complementary electrostatic field(Fig. 3), followed by an assignment of the atom coordinates and their types (Fig. 4).

The finalization of the ligand structure proceeds by discarding the outlying atoms, assigning the atom types and chemical bonds. This might result in several plausible structures. The ligand structures are then optimized with one of the semiempirical methods using a quantum chemistry package, and docked to the target protein. Their free energy of binding (ΔG) is calculated and compared to that of the known ligands of the protein.
The finalized galectin inhibitor (Fig. 5) appeared to be a completely new scaffold (as judged from the SciFinder search). Its estimated affinity (-23.8 kJ/mole) was lower than that for galectin’s known inhibitor lactose (-20.7 kJ/mole). Five derivatives of that ligand were constructed using published QSAR data. The best of those inhibitors had estimated ΔG of -32.1 kJ/mole, which is lower than ΔG of any of the published galectin-3 inhibitors.