“We’ve got this structure. What’s next?”
The target-to-hit discovery process at XR Pharmaceuticals looks as following:
1. Problem setup (with the client). Identification of the target, binding site, desired pKd, and the validation protocol.
|
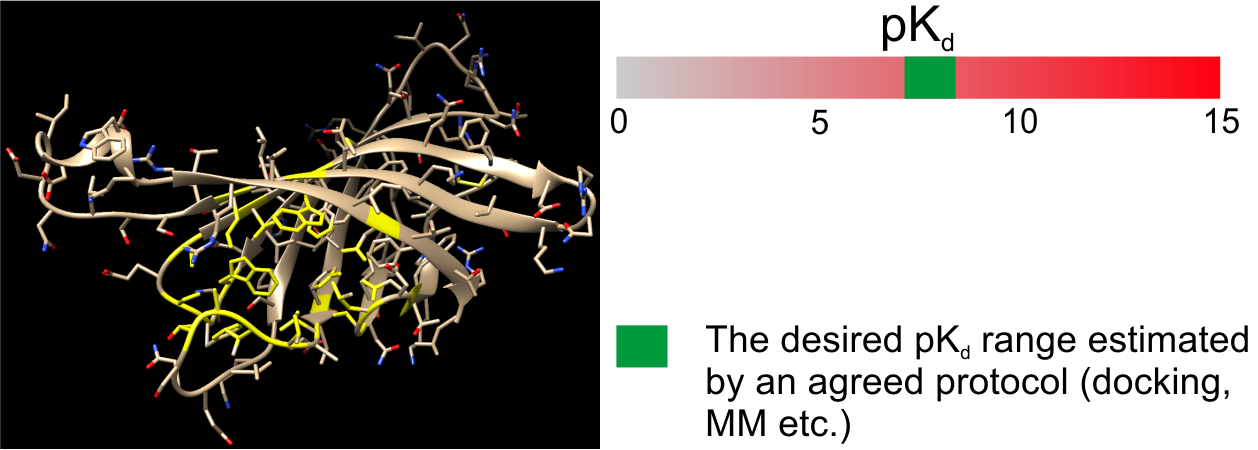

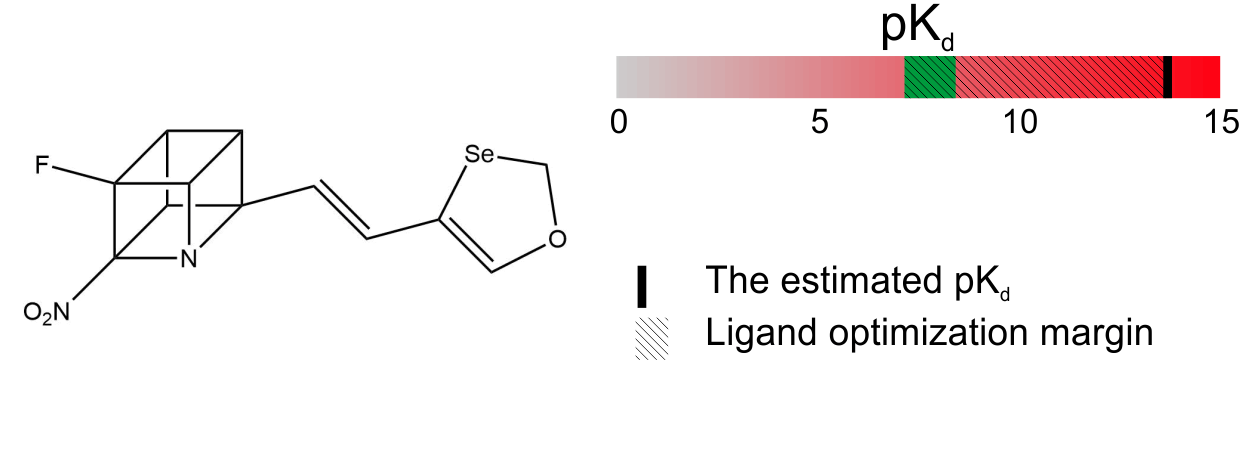
2. Ligand structure calculation. Estimation of the pKd. If the ligand passes the validation, the structure becomes the property of the client and all further steps are conducted by them.
|
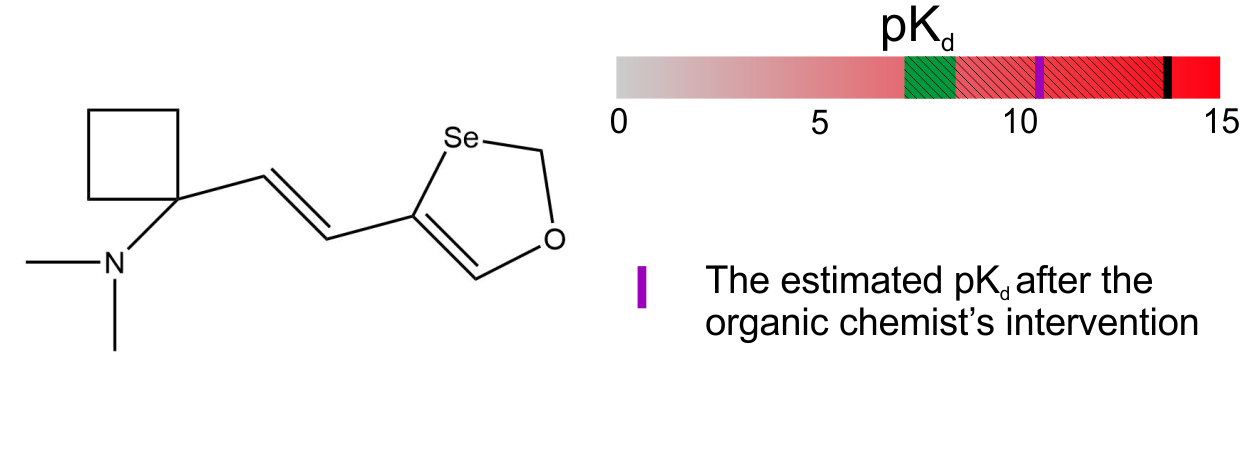
3. Client’s organic chemist opinion:
|
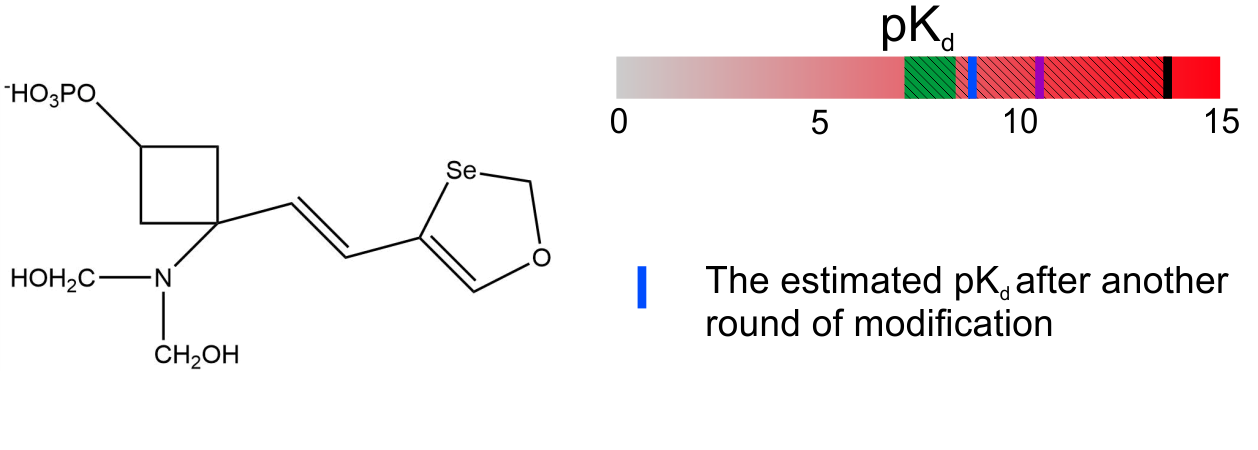
4. Client’s biomedical chemist opinion:
|

5. Is everybody happy?
|

Bottom line: our algorithm is aiming at delivering the structure with the highest theoretically possibly affinity. This lets the client to modify the structure wildly, while still meeting expectations on the required affinity.